当前,Coding Agents 在软件工程领域一路高歌猛进,科学家们看到此场景,也不禁寄予厚望:AI 智能体何时能以同样的速度,帮人类攻克药物设计、病毒监控与生物学建模的重重难关?
然而,一个残酷的现实是,AI 在生物学领域的发展要比编程领域慢得多……
近日,Anthropic 发表了一篇新科学博客 ——《为生物学智能体铺平道路》(Paving the way for agents in biology),文章指出:阻碍生物学 AI Agent 爆发的瓶颈,根本不在于大模型底座的推理能力不够强,而在于人类现有的生物学数据基础设施实在是太落后了。
因此,如果希望 AI Agent 真正参与生物学研究,生物数据基础设施必须变得更适合 Agent 使用。

这篇文章由 Laura Luebbert 撰写,她是一位生物学家与机器学习研究员。

有意思的是,Laura Luebbert 透露,这篇博客是在 Karpathy 官宣加入 Anthropic 之前一周就完成的,因为文中部分内容涉及 Karpathy ,还担心 Anthropic 会不会觉得这篇「Karpathy 味」太重。没想到的是,就在她把初稿发给 Anthropic 的同一天,他们就官宣了……

下面,我们就来详细看一下,这篇文章是如何分析的?
现有生物数据基础设施,对 Agent 来说太难走了
作者用了一个很有意思的类比,让 AI Agent 去操作生物数据基础设施,有点像开车穿过一座在汽车出现前就建好的老城:这座城市也许很美,规划也有其用心之处,但里面到处是狭窄、曲折的街道,现代车辆难以顺畅通行。对应到生物数据领域,就是各种特有的文件格式、分散的数据库,以及一次性的检索脚本。
当然,你也可以试着给这座城市补上交通标志、停车场,甚至偶尔拓宽几条道路,但它最基本的布局仍然难以通行,原因在于它原本就是为另一种交通方式设计的。
相比之下,软件基础设施几乎天然就适合「汽车」,也就是 Agent 使用:铺好的道路、清晰的车道、标准化信号,以及支持从起点到终点快速通行的系统,也就是版本控制、文档清晰的 API 和包管理器。
因此,Coding Agent 的发展速度明显快于生物学 Agent。
软件领域通常具备结构化的数字工作流和可靠接口,而计算生物学中用于数据检索和验证的基础设施,往往脆弱、异构,并且高度依赖具体流程。对应的,用来操作这些基础设施的工具,也就不得不变得定制化,并且只适用于特定领域或特定假设。
此外,软件可以给出容易测试的结果,并能快速编译和验证。例如,一个 Agent 可以通过生成补丁来解决 GitHub issue,只要补丁通过项目测试,就能判断是否有效。但在生物学中,这种简单、可验证、同时又有意义的奖励信号并不多。
所以,生物学 Agent 的瓶颈不只在推理能力,也在于缺少一种广泛可用的确定性执行层,来支持对生物数据的查询。科学家可以很自然地表达自己的意图,比如「找到所有带有这个结构域的人类激酶,并拉取它们的结构」。但 Agent 往往缺少一条可靠路径,去访问那些包含所需信息的数据库。
在生物学和科学工作流中,即使很小的错误,也可能带来严重后果。比如,从错误的基因组版本中提取坐标,可能会让后续的生物学解释失效;无意中混用 RefSeq 和 GenBank 记录、把部分基因组当作完整基因组、混淆分节病毒的片段名称,或者因为元数据字段不一致而漏掉相关记录,也都可能造成同样的问题。
科研的美感和难点正在于此:细节往往极其关键。
因此,如果希望 Agent 真正帮助科学发现,就需要对生物数据基础设施进行建设。
Karpathy 关于 Web 开发的「吐槽」,和生物学 Agent 面临的是同一问题
作者认为,Agent 的需求与人类构建的工具之间存在错配,并不是生物学领域独有,只要把 Agent 放进那些完全围绕人类使用习惯设计的环境里,类似的摩擦就会出现。
几个月前,Karpathy 在一场关于 AI 时代软件开发的演讲时吐槽,他用 Vibe Coding 写了一个小型 Web 应用,但让它真正跑起来时,身份验证、支付、部署这些环节,让他花了一周时间在浏览器后台到处点击。
为此,Karpathy 感慨:「代码反而是最容易的部分!大部分工作都在浏览器里,靠点击完成。」麻烦的是「打开这个 URL,点击这个下拉菜单。」
结论是:我们必须为 Agent 重新构建这些流程。
这正是生物学研究人员早已长期面对的痛点:我们试图让智能系统在一套为人类点击浏览器而设计的环境中工作,而这个环境充满了异构信息、隐含约定和各种需要人工操作的流程。
案例研究:病毒学里的「点击税」
早在 AI Agent 出现之前,计算生物学家和遗传学家就已经开始开发传统计算生物学工具,试图缓解这个问题。Biopython、BioPerl、BioJulia、Entrez Direct、BioMart、gget 以及许多其他工作流库,都是为了把生物数据从浏览器界面中解放出来,让研究人员可以直接对这些数据进行计算。
但问题在于,生物数据并不存放在统一数据库,也没有统一接口,更像一张混乱的道路网络:每条路都有自己的标识符、约定、格式、筛选逻辑和程序访问能力。有些数据可以很方便地通过程序调用,有些则困难得多。
病毒学则是难度较高的场景之一。从疫苗设计、诊断试剂开发,到为蛋白模型构建训练数据,很多研究工作流的第一步,都是从 NCBI Virus 中检索序列。NCBI Virus 是一个病毒序列记录集合,汇集了来自 GenBank、RefSeq 和国际 INSDC 生态系统的数据,其中也包括 Pathoplexus,并通过一个可搜索的网页界面提供访问。
参与病毒疫情监测工具建设的研究人员非常清楚,这些检索流程背后隐藏着多少专家知识。在病毒学实验室里,围绕 NCBI Virus 的数据集整理说明,常常是以一长串复杂筛选条件的形式流传。用户必须在网页界面中手动复现这些条件。
而这正是 Karpathy 所抱怨的那类「浏览器点击式工作流」。
文章以 2026 年 5 月中旬刚果宣告暴发的 Bundibugyo 埃博拉病毒疫情为例,说明了这一情况。
当一线研究人员测序出首批突发疫情的病毒基因组后,全球公共卫生官员需要立即回答三个迫在眉睫的问题:
这种新毒株与历史上的埃博拉病毒相比,变异有多大?
现有的诊断试剂盒还能准确把它检测出来吗?
现有的抗体药物和疗法是否依然能保护患者?
而要回答这些问题,分析的第一步必须是前往 NCBI Virus 数据库,将新基因组与历史数据进行比对。
然而,在病毒学实验室里,构建这种对照数据集的过滤条件非常复杂,往往作为长长的列表由科学家之间人肉传递。研究人员必须在复杂的 Web 界面中手动勾选数十个过滤器。对于人类来说这异常枯燥,而对于旨在通过自动化提升效率的 AI Agent 来说,简直是一场灾难……
Agent 自己尝试检索,会发生什么?
作者表示为了理解 Agent 和数据库之间的鸿沟,研究团队构建了一个基准测试 VirBench,包含 120 个真实风格的病毒序列查询任务,覆盖 40 种病原体,并配有人工验证的标准答案。任务来自病毒监测、诊断试剂设计、蛋白模型训练数据构建等实际场景。
比如其中一个任务要求 Agent 从 NCBI 检索 TaxID 3052462 对应的 Zaire ebolavirus 序列,并满足一系列条件:宿主是人类,采样地点在非洲,采样时间在 2014 年 1 月 1 日到 2014 年 6 月 20 日之间,序列长度至少 15200 个碱基,模糊字符 N 不超过 1900 个,并排除实验室传代样本。
当 Agent 独立完成这些查询时,结果差异很大。
Claude Sonnet 4、Claude Opus 4.7、Biomni、Edison Analysis、GPT-5.2-pro、GPT-5.5 的平均准确率从 16.9% 到 91.3% 不等。也就是说,前沿模型表现更好,但即便如此,也没有稳定达到可靠数据集构建所需的准确性和可复现性。
对这类任务来说,标准几乎必须接近 100%。因为漏掉或多拿一条记录,就可能影响诊断试剂是否覆盖当前流行病毒多样性,或者影响对疫情起点的判断。更麻烦的是,同一个模型在相同问题上重复运行三次,经常会给出差异很大的结果。
在上面的埃博拉病毒查询任务中,标准答案是 266 条序列,但 Claude Sonnet 4 三次运行分别返回了 106 条、15 条和 5 条。提示词完全相同,结果却高度不稳定。
这种不稳定会直接影响下游分析。研究团队用这些序列构建系统发育树,用来推断疫情中不同病毒样本之间的关系。其中一个重要指标是最近共同祖先时间,也就是 TMRCA。人工整理的数据集推断出的时间是 2014 年 1 月,和既有研究一致;但 Sonnet 4 检索出的部分数据集明显不完整,甚至有一次把共同祖先时间推回到 1922 年。

另一个例子涉及抗体疗法。研究人员检索埃博拉病毒糖蛋白序列,观察 maftivimab 和 MBP134 这类抗体药物靶向区域是否出现过突变。
结果显示,Sonnet 4 三次运行给出了三种不同印象:第一次接近人工查询结果,第二次漏掉大部分突变位点,第三次又强调了一组不同残基。
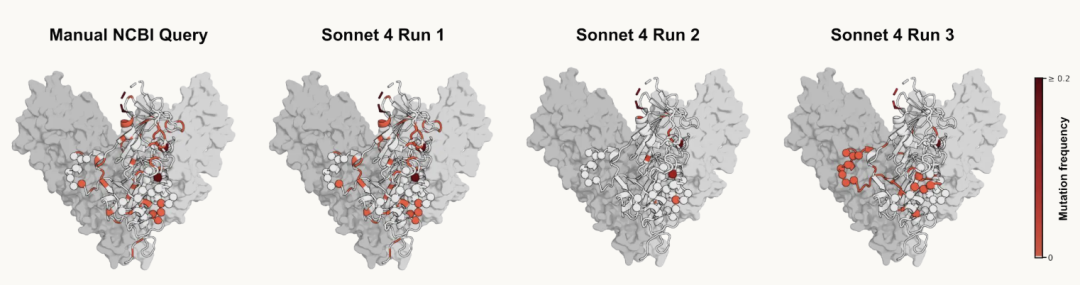
这说明,在科学研究中,看似只是检索细节的小差异,可能改变生物学结论。Agent 往往理解了任务,也愿意尝试执行,但缺少一种机器可操作、可验证、可重复的路径。最终答案可能看起来很合理,却是错的。
而这尤其危险,因为序列检索通常是更长时间生物工作流程的第一步……
gget virus:给病毒数据检索加一层确定性工具
为了解决这个问题,研究团队和 NCBI 研究人员合作开发了 gget virus,目标是把病毒数据检索变成 Agent 和人类都可以直接调用的稳定工具。
一开始,这似乎只是把几个 API 接起来,但实际情况复杂得多。NCBI Virus 是一个覆盖多个底层资源的门户,这些资源又分布在多个国家维护的国际同步序列数据库中。一个看似简单的查询,往往需要从多个地方拼接信息。
为了复现 NCBI Virus 网页界面的行为,gget virus 需要协调 REST、Datasets、E-utilities 等不同 API,它会判断哪些筛选条件可以通过现有 API 完成,哪些必须在本地检查,因为网页界面提供的一些筛选逻辑,并没有暴露在单一程序接口中。
它还会处理批量检索,确保 SARS-CoV-2、甲型流感这类大规模数据集被完整取回,而不会因为分页或中途截断而漏掉记录。如果筛选条件依赖另一个数据库里的补充信息,比如 GenBank 记录中某个序列是否包含特定病毒蛋白,gget virus 会取回这些记录,用它们完成过滤,并把相关 GenBank 信息保存在最终输出中。
最终,gget virus 输出的是人和机器都能读取的标准化结果,并带有详细日志,说明结果是如何产生的。这样一来,Agent 给出的答案不再只是「看起来合理」,而是可以检查、复现和审计。
加入 gget virus 后,所有 Agent 的准确率都提升到了 90% 以上,GPT-5.5 最高达到 99.7%。多次运行之间的波动也基本消失,不同模型之间的性能差距明显缩小。也就是说,一个确定性的检索层,让模型选择变得没有那么关键。

这一点很重要。可靠的数据集构建不应该依赖最新、最贵的模型,也不应该依赖研究人员知道哪个模型最适合哪个数据库。更便宜的模型加上合适工具,也可以减少不稳定性,让更多人获得可靠能力。
真正的启示:科学 Agent 需要「无聊但可靠」的底座
在文章的最后,作者强调,模型在生成假设、设计实验、推理机制时应该有创造力,但支撑这些创造力的底层部分,比如基因标识符、schema、检索逻辑、坐标系统、元数据约定、数据访问路径,必须足够稳定、确定、可复现。
gget virus 只是一个例子,未来更大的方向,是为生物数据构建一类「上下文引擎」:可靠、可被 Agent 访问的数据基础设施。类似探索也已经出现在 ToolUniverse、Edison Scientific 的 Robin、Biomni 以及其他生物医学 Agent 系统中。
当然,作者也承认,如果沿着上面实验结果所呈现出的模型能力曲线继续往前推,可以想象,在不远的未来,像gget virus这类工具可能会显得「没那么必要」:Agent 变得足够强,能够自己穿过混乱的门户网站、协调不同标识符、正确处理分页,并从失败中恢复过来……到了那个时候,工具框架也许就不再是必需品。
但即便 Agent 能够做到,也不意味着每一次都应该让它来处理,并且重新发明一遍流程。一个模型也许可以硬闯复杂混乱的生物信息学工作流,但对于日常科研工作来说,这种方式仍然可能太贵、太慢、太难审计,也太难让人放心。
而且,即便未来 Agent 真的让今天这些工具框架变得过时,这个教训对生物数据库依然成立:当我们思考用户是谁时,必须把 Agent 纳入考虑;当我们建设系统时,也必须面向规模化使用来设计……
更多内容可查看文章原文了解!
参考链接:
https://x.com/AnthropicAI/status/2064054837294354677
https://www.anthropic.com/research/agents-in-biology
https://x.com/NeuroLuebbert/status/2064055392016212080

© THE END
转载请联系本公众号获得授权
投稿或寻求报道:liyazhou@jiqizhixin.com