作者 | 论文团队
编辑 | ScienceAI
在药物设计、新材料开发等领域,精确模拟分子行为至关重要。传统的机器学习力场将分子视为由原子核和化学键构成的“骨架”,却忽略了真正决定分子性质的“灵魂”——电子。
电子密度,这一量子化学中的核心物理量,描述了电子在空间中的分布概率,从根本上决定了分子的能量、反应活性等所有性质。然而,通过量子化学计算获取电子密度数据成本极高,严重阻碍了该方向的研究。
近日,湖南大学/国家超算长沙中心联合宁波东方理工大学、香港科技大学(广州)AIMS Lab、华东师范大学、中国科学技术大学等团队在国际顶会NeurIPS 2025(神经信息处理系统大会,AI领域顶会)上发布研究成果 ——大规模电子密度泛函数据集EDbench。
这个涵盖336 万分子的“电子密度数据库”,首次填补了“电子级分子建模”的大规模数据空白,让AI预测分子性质的精度和效率实现双重突破,有望加速新药研发、新材料设计等关键领域的进程。

论文主页:https://hongxinxiang.github.io/projects/EDBench/
为什么电子密度是分子建模的“终极密码”?
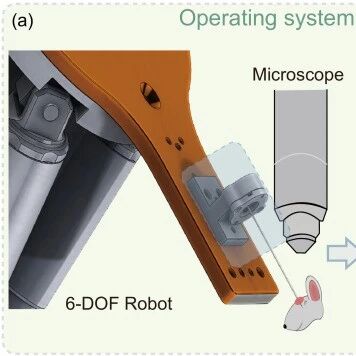
在量子化学中,分子的一切性质都藏在“电子分布”里,这是Hohenberg-Kohn 定理的核心结论。打个比方:如果把分子比作“智能手机”,传统AI分子建模(MLFFs)只关注“硬件零件(原子)”的排列,却忽略了“操作系统(电子)”的逻辑;而电子密度(ED)就是“操作系统的源代码”,能直接解锁分子的“底层功能”。

图(a)展示了从原子级到电子级的MLFF范式升级,其中电子密度(ED)是描述电子空间分布的量子力学中的基本物理量,可以提供分子系统更细致的物理表示,但是长期以来,获取ED数据堪称“两难”:
实验测量(如X射线衍射)依赖千万级设备,单种分子的ED数据需数月才能获取;
理论计算(DFT)虽通用,但算一个小分子要245秒,想攒“百万级数据集”需单核心连续计算23年。
这就导致AI分子建模始终卡在“原子级”——比如预测药物分子与靶点的结合活性时,传统模型只能通过“原子间距”间接推断,却看不到“电子富集区”的真实作用,精度往往差强人意。
EDBench数据集:规模与质量的双重突破
本论文中,EDBench的建成正好解决了该领域长期存在的“数据荒”问题,该数据集的创建并不是“简单攒数据”,而是“精度、规模、实用性”的多重考量。
1.规模突破:336 万分子的“算力攻坚”
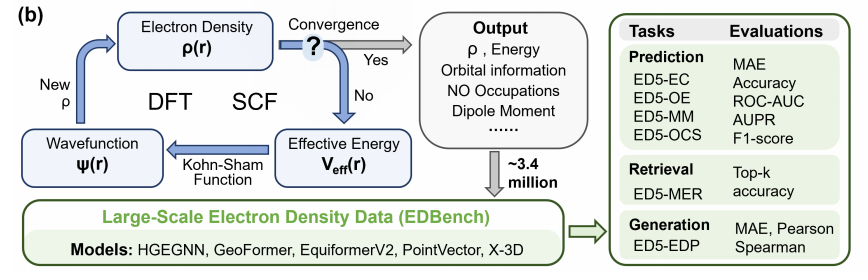
从细节角度来看,图(b)展示了数据集构建中的DFT计算流程,该流程是通过自洽场(SCF)迭代来实现的,从宏观角度来看,EDBench基于PCQM4Mv2数据集构建,涵盖3,359,472个类药分子,规模超同类成果:
材料领域经典ED数据MP仅含12万条数据,且无量子性质标注;
药物领域最大电子相关数据集∇²DFT 虽有190万条,但不含直接ED分布(仅密度矩阵)。
更重要的是,EDBench输出的是CUBE格式的3D电子密度云图——相当于给每个分子拍了“电子CT”,能直接看到电子在原子周围的富集程度。
2.精度控制:从泛函到基组的“定制化方案”
为了让ED数据符合真实量子规律,团队基于图(c)中Jacob’s ladder做了三重优化:

泛函选择:选用B3LYP混合泛函——这是量子化学的“万金油”,在合成化学、药物研发等领域经过验证,能平衡电子相关性描述与计算效率;
基组适配:普通分子用6-31G基组,含硫分子追加6-31+G基组——因为硫原子的电子离域性强,普通基组会低估其ED分布;
自旋适配:自动切换RHF/UHF参考波函数——闭壳层分子(如甲烷)用RHF,开壳层分子(如氧分子)用UHF,避免自旋污染导致的误差。
3.标注维度:AI 建模的“全要素养料”
EDBench不止提供ED数据,还同步输出6类能量成分(核排斥能、交换关联能等)、7种轨道能量(HOMO-2 至 LUMO+3)、4类偶极矩(X/Y/Z 分量及模长)。这种“多维度标注”正是AI突破“单任务拟合”的关键——例如,模型可通过“ED分布→轨道能量→反应活性”的链式学习,实现更本质的性质预测。
三大基准任务:验证AI的“电子理解能力”
为避免数据集“只存不用”,团队设计了覆盖ED核心应用场景的基准任务,均采用scaffold split(OOD泛化性测试),直击AI建模的真实痛点。
1. 量子性质预测:ED比原子特征强在哪?
4个子任务中,X-3D(点云模型)的表现印证了ED的优势:
ED5-OE(轨道能量预测):HOMO能量MAE仅0.0198 eV,较原子级模型(MAE≈0.03 eV)精度提升34%——因为轨道能量与局部ED分布直接相关,AI 能快速捕捉“电子富集区”的微观特征;
ED5-OCS(开/闭壳层分类):AUPR达61.54%,解决了传统模型“分不清电子态”的核心难题——开壳层分子(如自由基)的ED分布呈“不对称性”,AI 能通过这种特征精准分类。
2. 跨模态检索:“结构-密度”双向对齐的突破
ED5-MER 任务中,模型需实现“分子结构→ED”和“ED→分子结构”的双向检索,这对药物虚拟筛选、材料逆设计至关重要。

图中清晰展示:EquiformerV2(分子结构编码器)+X-3D(ED 编码器)的组合表现最优,ED→MS 检索Top-1准确率达 78.71%,MS→ED 达 78.36%。这意味着:给一张实验测得的电子云图,AI 能精准匹配对应的分子结构;反之,输入分子结构也能快速生成ED分布,为“电子驱动的逆设计”奠定基础。
3. ED 生成:1.8 万倍提速的“DFT 平替方案”
团队提出HGEGNN 异构图模型(将原子与电子作为双类型节点),输入分子结构即可生成ED:
速度:0.013秒/分子,较DFT(245 秒/分子)提速18907倍;
质量:生成ED的Pearson相关达99.2%,且用于下游能量预测时,MAE较DFT原始数据降低11%(209.29→186.38)——AI生成的ED因过滤了DFT计算的噪声,反而更适合模型学习。
生成 ED 的质量“反超”DFT?
上述HGEGNN异构图模型引出了一个颠覆性发现:AI生成ED数据,在下游任务中表现竟优于DFT原始数据。
团队用HGEGNN生成3组ED数据(G、G、G),替换ED5-EC任务中的DFT数据,训练PointVector模型后发现:生成数据的平均MAE从209.29降至186.38,相对提升11%。

图中蓝色柱DFT数据的模型表现,绿色柱为AI生成数据的表现——这并非说明 AI生成的ED“更符合物理真实”,而是其“更平滑的分布”更契合深度学习模型的归纳偏好,减少了DFT计算中的数值噪声,为“高效-高精度建模”提供了新思路。
参数玄机:阈值与采样点如何影响性能?
ED数据的“高维度”(百万级采样点)可能导致模型效率低下,团队通过消融实验找到最优参数。
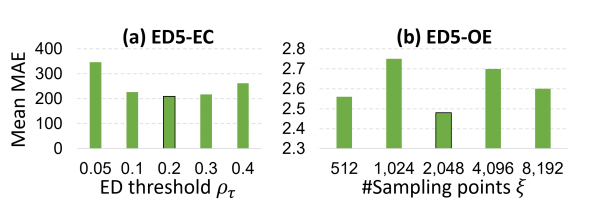
左图(a)显示:随着ED阈值ρτ从0.05增至0.2,PointVector在ED5-EC任务的 MAE从346.36降至209.29;但ρτ超过0.2后,性能反而下降——因为过高的阈值会过滤掉化学键附近的低密度电子(化学活性区),这提示:ρτ=0.2 是“信息保留与效率”的黄金平衡点。
对 AI for Science 的启示:从“拟合”到“理解”
EDBench 的价值远超“一个数据集”:它首次证明了“电子级表征”在AI建模中的可行性,为几何深度学习(GDL)开辟了新方向——过去GDL聚焦“原子间对称关系”,现在可转向“电子密度的空间连续性”建模。
目前,EDBench的全部数据(含CUBE格式电子云图)、代码及可视化工具已通过哈佛Dataverse开放:
https://dataverse.harvard.edu/dataverse/EDBench
正如团队通讯作者所言:“EDBench”不是终点,而是让AI“看懂电子”的起点——当模型能真正理解电子分布,新药研发、新材料设计才有可能从“试错”走向“预测”。
人工智能 × [ 生物 神经科学 数学 物理 化学 材料 ]